Introduction
La calorimétrie différentielle à balayage (DSC) est l'une des méthodes d'analyse thermique les plus fréquemment utilisées pour le contrôle de la qualité. Sa grande popularité n'est pas seulement due au fait qu'elle fournit des informations substantielles sur les propriétés des matériaux telles que la transition vitreuse, la Températures et enthalpies de fusionL'enthalpie de fusion d'une substance, également connue sous le nom de chaleur latente, est une mesure de l'apport d'énergie, généralement de la chaleur, nécessaire pour convertir une substance de l'état solide à l'état liquide. Le point de fusion d'une substance est la température à laquelle elle passe de l'état solide (cristallin) à l'état liquide (fusion isotrope). fusion ou la conversion cristal-cristal, mais aussi à sa facilité et à sa rapidité d'utilisation. En particulier, tous les DSC NETZSCH offrent la possibilité d'automatiser la plupart des étapes de mesure, de sorte que l'évaluation et même l'identification d'un matériau peuvent être effectuées automatiquement.
Expérimental
Toute mesure DSC sur des polymères doit comprendre trois séries de mesures consistant en deux mesures de chauffage, entre lesquelles l'échantillon est refroidi à une vitesse contrôlée. Chaque courbe de mesure peut fournir des informations différentes sur l'échantillon.
- La première série de mesures de chauffage fournit des informations sur l'historique thermique de l'échantillon. Par exemple, à quelle vitesse a-t-il été refroidi pendant le traitement ? Quelles étaient les conditions de température et d'humidité de stockage ? A-t-il été soumis à des contraintes mécaniques ?
- En refroidissant l'échantillon dans des conditions définies (vitesse de refroidissement, atmosphère), on obtient un historique thermique connu.
- Le (second) chauffage qui suit est utilisé pour déterminer les propriétés de l'échantillon, ce qui est particulièrement important si plusieurs polymères doivent être comparés, par exemple dans le cadre d'un contrôle de qualité.
Cependant, l'étude suivante montre que le segment de refroidissement, souvent négligé, peut également présenter un grand intérêt. Des mesures ont été effectuées sur deux échantillons de PEEK non remplis et étudiés au moyen de la DSC. Le tableau 1 résume les conditions des mesures DSC effectuées sur les deux échantillons.
Tableau 1 : Conditions d'essai pour les mesures DSC
Échantillon 1 | Échantillon 2 | |
|---|---|---|
| Appareil | DSC 214 Polyma | |
| Masse de l'échantillon | 12.05 mg | 5.57 mg |
| Plage de température | 30°C à 400°C (deux fois) | |
| Vitesses de chauffage et de refroidissement | 10 K/min | |
| Atmosphère | Azote (40 ml/min) | |
| Creuset | Concavus® (aluminium), fermé par un couvercle percé | |
Résultats des mesures
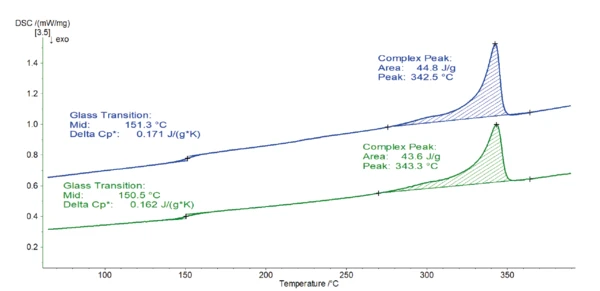
La figure 1 présente les résultats de la deuxième série de mesures de chauffage normalement utilisée pour une telle analyse.
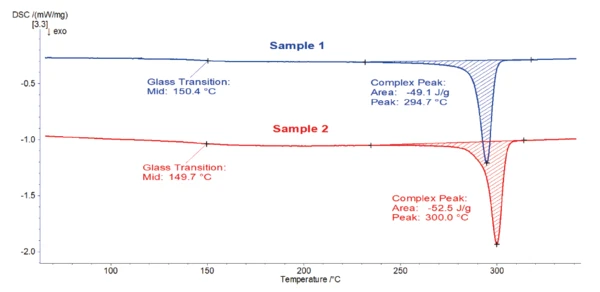
Les deux courbes sont très similaires. L'étape EndothermiqueUne transition d'échantillon ou une réaction est endothermique si la conversion nécessite de la chaleur.endothermique détectée à 150-151°C résulte de la transition vitreuse du polymère. Le pic suivant, situé entre 270°C et 360°C, est dû à la Températures et enthalpies de fusionL'enthalpie de fusion d'une substance, également connue sous le nom de chaleur latente, est une mesure de l'apport d'énergie, généralement de la chaleur, nécessaire pour convertir une substance de l'état solide à l'état liquide. Le point de fusion d'une substance est la température à laquelle elle passe de l'état solide (cristallin) à l'état liquide (fusion isotrope). fusion de la phase cristalline. Pour les deux échantillons, la température maximale se situe à 343°C et est associée à une enthalpie de Températures et enthalpies de fusionL'enthalpie de fusion d'une substance, également connue sous le nom de chaleur latente, est une mesure de l'apport d'énergie, généralement de la chaleur, nécessaire pour convertir une substance de l'état solide à l'état liquide. Le point de fusion d'une substance est la température à laquelle elle passe de l'état solide (cristallin) à l'état liquide (fusion isotrope). fusion de 44-45 J/g. Ce pic de Températures et enthalpies de fusionL'enthalpie de fusion d'une substance, également connue sous le nom de chaleur latente, est une mesure de l'apport d'énergie, généralement de la chaleur, nécessaire pour convertir une substance de l'état solide à l'état liquide. Le point de fusion d'une substance est la température à laquelle elle passe de l'état solide (cristallin) à l'état liquide (fusion isotrope). température de fusion est typique du PEEK [1].
Sur la base de ces courbes de chauffe, il n'y a pas de différence notable entre les échantillons 1 et 2. Le contrôle de qualité conclurait qu'il s'agit du même matériau.
S'agit-il du même matériau ? La réponse vient de la rhéologie
La rhéométrie rotative permet d'obtenir plus d'informations sur ces échantillons. Le polymère fondu est placé entre les plaques de mesure du rhéomètre rotatif Kinexus. Les propriétés viscoélastiques de l'échantillon sont déterminées par l'oscillation de la géométrie supérieure à une fréquence et une amplitude spécifiées.
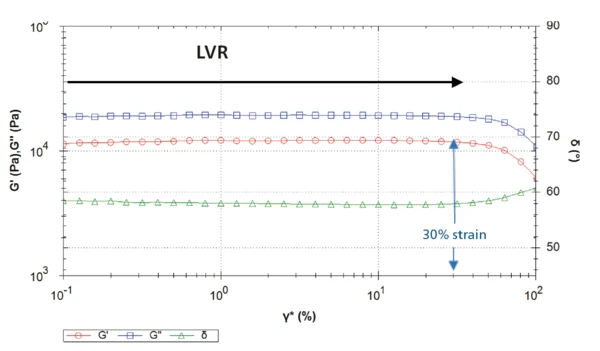
Un balayage de fréquence a été effectué sur les deux polymères, en veillant à ce qu'il soit réalisé dans la Région viscoélastique linéaire (LVER)Dans le LVER, les contraintes appliquées ne sont pas suffisantes pour provoquer une rupture de la structure, ce qui permet de mesurer d'importantes propriétés micro-structurelles. région viscoélastique linéaire (LVR) de chaque échantillon (voir l'encadré). Un balayage d'amplitude sert de mesure préliminaire pour déterminer la limite de la LVR de l'échantillon.
Le tableau 2 détaille les conditions des balayages d'amplitude et de fréquence.
Tableau 2 : Conditions d'essai pour les mesures d'oscillation
Balayage d'amplitude | Balayage de fréquence | |
|---|---|---|
| Appareil | Kinexus ultra+ avec chambre chauffée électriquement (EHC) | |
| Géométrie | PP25 (plateau-plaque, diamètre : 25 mm) | |
| Température | ||
| Déformation par cisaillement | de 1 % à 100 % | - |
| Contrainte de cisaillement | - | 1000 Pa (échantillon 1) ; 500 Pa (échantillon 2) |
| Fréquence | 1 Hz | 0.01 Hz à 20 Hz |
| Atmosphère | Débit d'azote ( 1 l/min) | |
LVR - Gamme viscoélastique linéaire
La LVR est la plage d'amplitude où la déformation et la contrainte sont proportionnelles. Dans la LVR, les contraintes (ou déformations) appliquées ne sont pas suffisantes pour provoquer une rupture structurelle et, par conséquent, les propriétés microstructurelles sont mesurées.
La figure 2 illustre les courbes résultant du balayage d'amplitude sur l'échantillon 1. Pour une déformation de cisaillement allant jusqu'à environ 30 %, le module de cisaillement élastique G' reste constant. Par conséquent, les déformations de cisaillement supérieures à 30 % seront destructrices pour ces échantillons, car ils se trouvent en dehors de la LVR. La déformation de cisaillement à 30 % correspond à une contrainte de cisaillement d'environ 10 000 Pa.
Par conséquent, une contrainte de cisaillement sélectionnée de 1 000 Pa pour les mesures oscillatoires ultérieures sur ces échantillons, telles qu'un balayage de fréquence, se situe à l'intérieur de la LVR et n'est donc pas destructive.
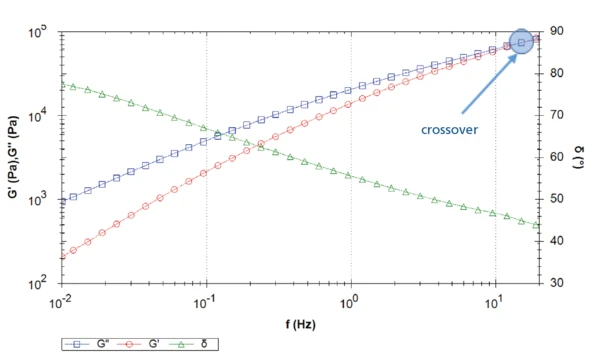
La figure 3 illustre les courbes des modules de cisaillement élastique et de perte, ainsi que l'angle de phase capturé pendant le balayage de fréquence. Dans la direction des basses fréquences, le Module visqueuxLe module complexe (composante visqueuse), module de perte ou G'', est la partie "imaginaire" du module complexe global des échantillons. Cette composante visqueuse indique la réponse liquide ou déphasée de l'échantillon mesuré. module visqueux domine le module élastique (angle de phase > 45°) : Le matériau est un liquide viscoélastique. Un croisement est constaté à une fréquence d'environ 15 Hz : Pour les fréquences plus élevées (c'est-à-dire les échelles de temps courtes), les propriétés "solides" du matériau dominent le comportement.
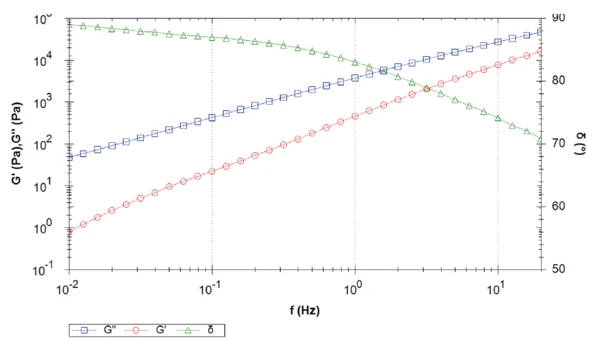
La figure 4 montre le balayage de fréquence de l'échantillon 2. Sur l'ensemble de la mesure, le module de cisaillement visqueux domine le module de cisaillement élastique, ce qui se traduit par un angle de phase supérieur à 45°. L'angle de phase diminue avec l'augmentation de la fréquence. En d'autres termes, à basse fréquence (ou à longue échelle de temps) dans la matière fondue, l'échantillon se comporte presque comme un fluide visqueux pur (angle de phase proche de 90°) avec des propriétés élastiques minimales.
Dans cette gamme de fréquence mesurée, aucun croisement n'est détecté. Le croisement se produit à une fréquence supérieure à la gamme de fréquences mesurée, c'est-à-dire supérieure à 20 Hz. Plus la fréquence du croisement est élevée, plus la masse moléculaire est faible [2]. Les deux matériaux diffèrent apparemment par leur poids moléculaire, ce qui n'a pas pu être observé dans les transitions de Températures et enthalpies de fusionL'enthalpie de fusion d'une substance, également connue sous le nom de chaleur latente, est une mesure de l'apport d'énergie, généralement de la chaleur, nécessaire pour convertir une substance de l'état solide à l'état liquide. Le point de fusion d'une substance est la température à laquelle elle passe de l'état solide (cristallin) à l'état liquide (fusion isotrope). fusion de la DSC.
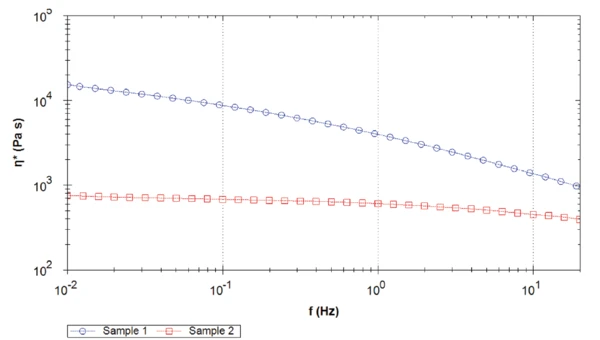
La figure 5 compare la viscosité complexe des deux échantillons. Pour toute la gamme de fréquences mesurée, l'échantillon 1 présente une viscosité complexe plus élevée que l'échantillon 2, avec plus d'une décennie de différence à 0,1 Hz. En outre, l'échantillon 2 de PEEK atteint un plateau newtonien autour de 1 Hz. Au contraire, la viscosité complexe de l'échantillon 1 continue d'augmenter à mesure que les fréquences diminuent.
La différence entre les valeurs du plateau de la viscosité complexe est due aux différentes masses moléculaires. Plus la masse moléculaire est élevée, plus le plateau de la viscosité à cisaillement nul est élevé [2].
Remarque : ici, c'est la viscosité complexe, et non la viscosité de cisaillement, qui est déterminée. Cependant, selon la règle de Cox-Merz, les deux valeurs peuvent être assimilées [3].
La viscosité complexe, ŋ*, est obtenue à partir de la rigidité complexe, G*, et de la fréquence angulaire, ω. ŋ* = G*/ω Elle est exprimée en [Pa-s].
La figure 6 illustre les courbes de refroidissement DSC des deux matériaux PEEK. Le pic ExothermiqueUne transition d'échantillon ou une réaction est exothermique si elle produit de la chaleur.exothermique détecté entre 310°C et 240°C provient typiquement de la CristallisationLa cristallisation est le processus physique de durcissement au cours de la formation et de la croissance des cristaux. Au cours de ce processus, la chaleur de cristallisation est libérée.cristallisation du PEEK. Les températures de transition vitreuse ont été détectées autour de 150°C. Une observation intéressante est la différence entre les températures de CristallisationLa cristallisation est le processus physique de durcissement au cours de la formation et de la croissance des cristaux. Au cours de ce processus, la chaleur de cristallisation est libérée.cristallisation maximales (Tc), le matériau de plus faible poids moléculaire (échantillon 2 de PEEK) présente une Tc inférieure de5°C .
Alors que la différence de poids moléculaire des deux polymères PEEK n'a pas d'influence sur leurs pics de Températures et enthalpies de fusionL'enthalpie de fusion d'une substance, également connue sous le nom de chaleur latente, est une mesure de l'apport d'énergie, généralement de la chaleur, nécessaire pour convertir une substance de l'état solide à l'état liquide. Le point de fusion d'une substance est la température à laquelle elle passe de l'état solide (cristallin) à l'état liquide (fusion isotrope). fusion, ils présentent des comportements de refroidissement différents ; plus le poids moléculaire est faible, plus la température de CristallisationLa cristallisation est le processus physique de durcissement au cours de la formation et de la croissance des cristaux. Au cours de ce processus, la chaleur de cristallisation est libérée.cristallisation est élevée. Alors que le cycle de refroidissement dans le DSC peut indiquer, mais pas prédire à lui seul, la différence de poids moléculaire, la mesure rhéologique fournit clairement cette information.
Conclusion
La calorimétrie différentielle à balayage est une technique bien connue et facile à utiliser, qui permet une analyse rapide des propriétés thermiques des polymères. Les évaluations de contrôle de la qualité sont généralement effectuées sur les deuxièmes courbes de chauffage DSC. Dans certains cas, le segment de refroidissement peut également s'avérer très utile. La rhéométrie est une technique complémentaire qui fournit des informations sur la viscosité et les propriétés viscoélastiques des matériaux. La combinaison de la DSC et de la rhéométrie permet d'obtenir un aperçu beaucoup plus approfondi des propriétés du matériau par rapport aux informations fournies par une seule méthode.